
In recent years, Poland has become an attractive location in the EU for conducting clinical trials, owing to the availability of state-of-art infrastructure, medically qualified personnel, and legislative policies fostering healthcare research. Moreover, Poland is also considered to be one of the fast-growing markets for medicines and healthcare (PWC: From Vision to decision: Pharma 2020). In this series of articles, we recapitulate the clinical trial process with an aim to highlight the services offered to the pharmaceutical industry and medical devices companies by Advanced Clinical Trials (ACT), a Polish clinical research organisation (CRO) with over 20 years of experience in clinical trial management.
CHAPTER ONE
THE CLINICAL TRIAL PROCESS: DRUG DEVELOPMENT
Since the first randomised clinical trial was conducted in the 1940s, the process of drug development has been revised and refined to address the paramount issues of safety and efficacy of emerging treatments. In the subsequent »70 years, the definition of ‘drug’ has also evolved in parallel. While drug development in the past focussed primarily on novel chemical entities (NCEs), in the present day such a term could also be applied to biologics, stem cell therapies, gene-based therapies, and a whole host of other treatment modalities. In the European Union (EU), these new treatments which are still in the clinical evaluation phase are termed as investigational medicinal products (IMPs).
What has essentially remained unchanged are the basic principles underlying the clinical evaluation process. Generally speaking, this can be divided into two parts – the preclinical (non-clinical) phase which involves drug discovery, and the clinical phase which entails drug development (Fig. 1).
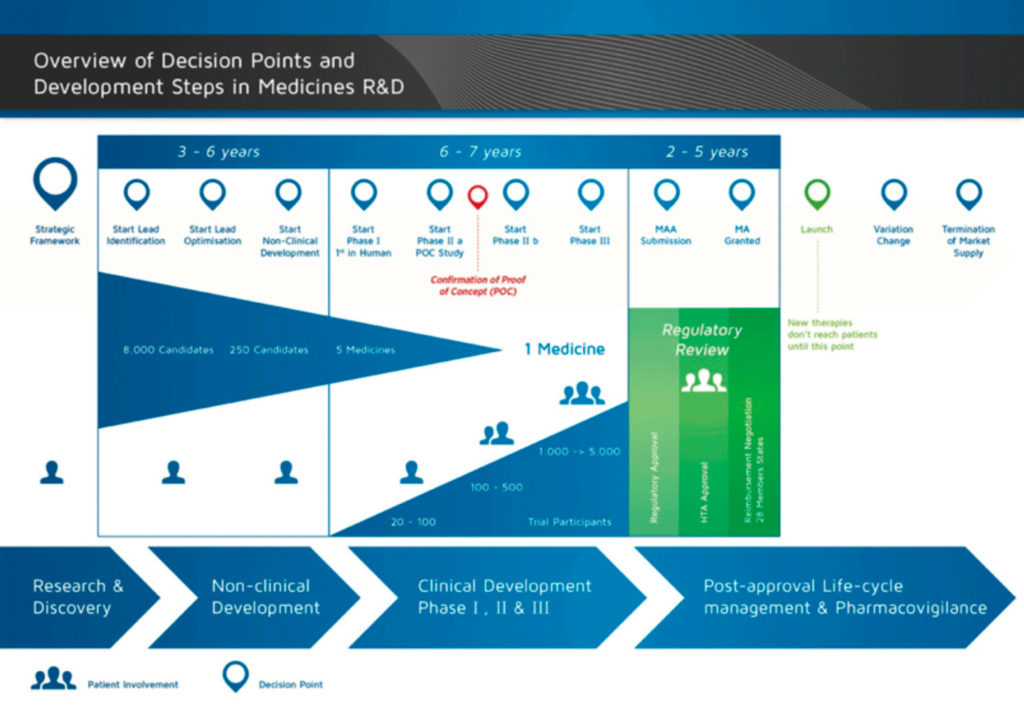
Using NCEs as an example of an IMP, the clinical phase can be further divided into four stages:
Phase 1– First-in-human studies, designed to assess safety and tolerability, pharmacokinetic profile of the IMP in humans, and estimation of a safe dose range for the IMP. Phase 1 studies are usually conducted in healthy volunteers. The cohort size ranges from 20-100 subjects.
Phase 2 – Studies designed to establish the ’proof-of-concept’ of efficacy of IMPs and reiterate human safety in comparison with either placebo controls or standard-of-care. Phase 2 studies are conducted in 100-500 subjects presenting the disease for which the IMP will be indicated.
Phase 3– Studies designed to recapitulate the safety and efficacy observed during Phase 2 trials in a larger patient population (1000-5000 subjects) and conducted across multiple centres/countries. Aimed at demonstrating the superiority/non-inferiority/equivalence (generic drugs), of IMPs to standard of care.
Phase 4– Studies conducted as a part of post-marketing surveillance, once the IMP has received a marketing approval. Assist in capturing various key data from the real-world scenario such as safety profile in a larger population or sub-populations, patient/physician acceptance/satisfaction, rare adverse events undetected during Phase 1-3 trials, etc.
Clinical trials can also be classified based on the sponsor. Commercial trials are those which are sponsored by the pharmaceutical industry with a principal aim at bringing to market a new medicine. On the other hand, non-commercial trials are generally sponsored by universities, medical institutions, patient organisations, etc. Non-commercial studies are generally performed on drugs already in the market, albeit for a specific set of patients or for testing different regimens or for a new indication. While the majority of clinical trials are commercial by nature, approximately 20%–40% (<10% in eastern Europe) are non-commercial. However, generally speaking, the requirements and procedures for application for conducting all clinical trials are the same.
A number of substantial changes in regulatory requirements for clinical assessment have been executed by the EMA in recent times e.g. transitioning from CTD 2001/20/EC to CTR 536/2014 (for the clinical trial process) and the implementation of MEDDEV 2.7.1 ver. 4 (for evaluation of medical devices). CROs such as ACTnot only help in leading the clinical studies both globally and locally, but also offer effective consultation to the pharmaceutical and medical device industry on how to proceed with regards to the EMA guidelines.
In conclusion, the clinical development of a new IMP is a process long and laborious and involves a substantial investment of time and money. To maximise the returns on this investment requires meticulous and insightful planning. In the next chapter, we will discuss how an appropriate study design ensures this.