
Ensuring compliance with regulatory requirements at local, national and EU level is a key element in the design of clinical trials in every EU country. We will write about upcoming changes in regulations in 2020 in this article.
While logistics and planning constitute the majority of the work involved in clinical trial design, a critical component of this stage is ensuring compliance with regulatory requirements at the local, national, and EU levels. Especially so since the clinical trial process will see a dramatic and progressive shift with new EU regulations being rolled out by as early as 2020. The two key ones that we shall consider in this chapter are the Clinical Trial Regulation (CTR) EU No. 536/2014 for investigational medicinal products and the Medical Devices Regulation (MDR) MDR 2017/745 for medical devices.
The main changes in the conduct of clinical trials
The changing landscape of conducting
clinical trials around the world and the intent to make the EU one of the hubs
for clinical research served as an impetus for introducing the CTR, especially
since the earlier Clinical Trial Directive (CTD) 2001/20/EU was found
to potentially be tedious and deterrent to this exercise. Thus, the main goals
of CTR 536/2014 were – (1) to ensure EU-wide harmonization of the approval
process and with predefined time limits; (2) to introduce a common evaluation process
for multinational trials; (3) to reduce the scope of regulatory autonomy at
member state level; (4) to make the EU competitive in research; (5) to generate
high-quality scientific data; and (6) to ensure patient safety. While the CTR
was adopted by the European Parliament in 2014, it is anticipated to come into
effect in the middle of 2020 with a further 3 years of a transition period till
the CTD ceases to be applicable (Figure
1).[1]
Once in effect, the CTR is expected to streamline the clinical trial
application process across Europe, ensure greater transparency in the clinical
processes and data sharing, and consequently enhance safety and efficacy of
drugs.
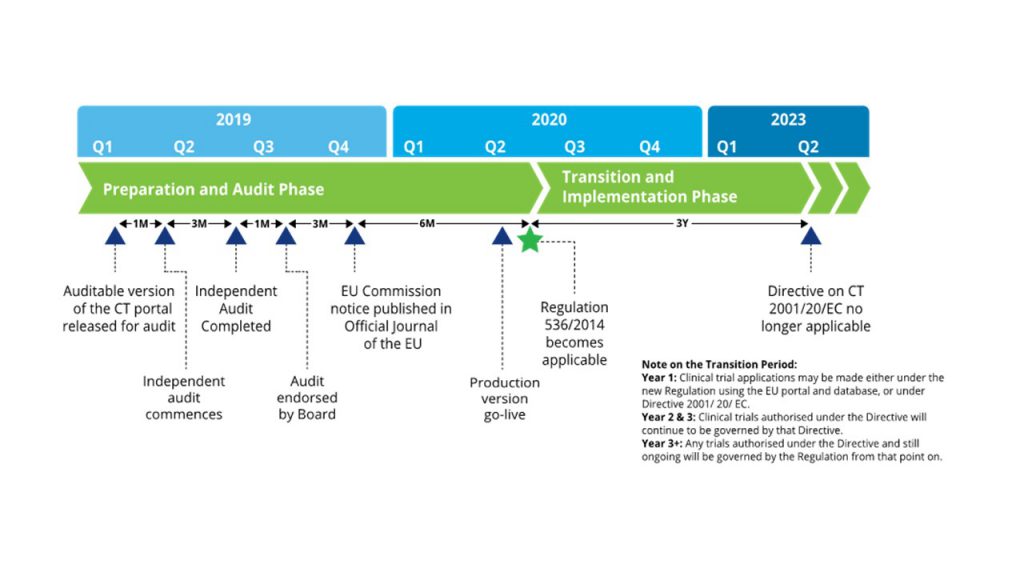
Figure 1. Timeline for CTR compliance. (Source: Deloitte)
One portal in EU for all clinical trials applications
In order to achieve these goals, the EU has now introduced a single portal linked to a European database through which all applications will be evaluated and decided upon. Other changes accompanying the CTR include focussing only on investigational medicinal products, lighter regulatory requirements for drugs with market authorisation, simplification of reporting requirement, and central approval at the EU level. At the time of writing this blog, we in Poland are awaiting the launch of the platform as well as national laws that will assist in executing the regulation.
Important patient safety
So how does CTR 536/2014 integrate the interests of various stake-holders? From a sponsor’s perspective, this new regulation simplifies the process immensely by way of a centralised procedure for submitting trial applications and evaluation of trial proposals. In addition, sponsors will also find it easier to further evaluate already approved medicinal products with an established safety profile since applications for such trials will have fewer requirements compared to the past. From a patient’s perspective, additional security provisions in the conduct of clinical trials as well as the establishment of a searchable trial database increases the amount of information available to the patient populace. Moreover, simplification of the informed consent process and inclusion of further safety measures assists trial investigators in recruiting subjects. The new regulation also allows for co-sponsoring of trials and efficient archiving of trial documents which helps CROs conduct trials smoothly on behalf of sponsors. Hence, CTR 536/2014 is expected to herald an era of a streamlined and efficient clinical trial process that will be both robust and transparent
Medical devices sector
In the interim, the European Commission has also overhauled and updated the regulatory oversight process on the exponentially growing medical devices sector with the release of EU MDR and EU IVDR, expected to come into effect latest by 2020 and 2022, respectively (Figure 2).[2]
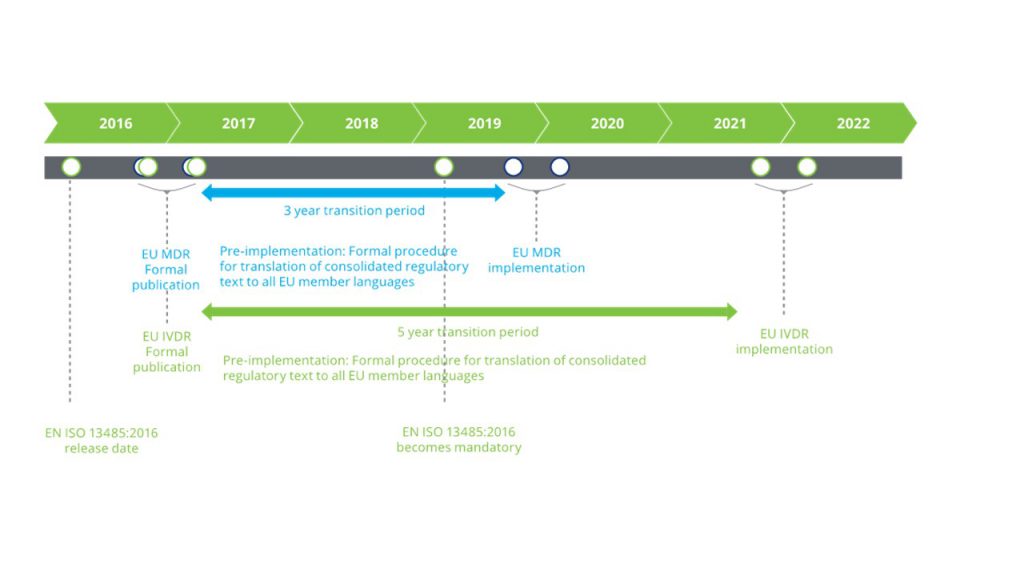
Figure 2. Timeline for MDR and IVDR compliance. (Source: Deloitte)
The main aim behind drafting new regulations was to ensure conformity of medical devices and improve their safety profile across the EU with an integrated process. In the clinical context, perhaps the most impactful change that will be implemented by the MDR will be on the issue of ‘clinical equivalence’.[3] While in the past this could be established through review of published literature, the stringency brought about by MDR means that for numerous Class IIa and IIb devices clinical investigations will be required in order to establish safety, efficacy, and ‘fitness for use’. Moreover, the MDR also increases the responsibilities of the Notified Bodies, institutions which evaluate the conformity of devices, as well as oversight on these. This is more than likely to increased scrutiny, vigilance, and audits on the device manufacturers.
The exact impact of MDR and IVDR is yet to be known. However, what is a given is that manufacturers will have to invest in reorganising their internal clinical and quality processes, reassessing their portfolios, and ensuring compliance.
As important are these new regulations in the due course of streamlining the regulatory processes in order to bring safer and more effective health interventions in the EU, these can be a bit of a challenge to negotiate for pharmaceutical companies or device manufacturers. CROs, by the very nature of the services they provide, are not only well-versed with existing regulatory requirement but also have their finger on the changing pulse of regulatory requirements. Thus, CROs will continue to occupy a key position in assisting their clients interested in conducting clinical research in the EU.
In the next chapter, we will discuss the
main focus of the clinical trial process – the investigational medicinal
product.
[1] Deloitte Touche Tohmatsu Limited. EU Clinical Trial Regulation: Building a successful programme. 2018. Available from: https://www2.deloitte.com/global/en/pages/life-sciences-and-healthcare/articles/clinical-trial-regulation.html
[2] Deloitte Touche Tohmatsu Limited. Preparing for the future: The new European Union medical devices regulation. 2018. Available from: https://www2.deloitte.com/content/dam/Deloitte/global/Documents/Life-Sciences-Health-Care/gx-eu-med-device-regulation.pdf
[3] Jaap Laufer. Understanding EU clinical investigation requirements for devices. 2017. Available from: https://www.emergobyul.com/resources/articles/white-paper-clinical-evaluation-review-ce-marking